gromacs gmxMMPBSA及能量景观图学习笔记
gmxMMPBSA快速操作笔记
1. 安装
wget https://valdes-tresanco-ms.github.io/gmx_MMPBSA/dev/env.yml
conda env create --file env.yml
conda activate gmxMMPBSA
#💬 Install mpi4py and AmberTools
conda install -c conda-forge "mpi4py=4.0.1" "ambertools<=23.3" -y -q
#💬 Install dependencies for ploting
conda install -c conda-forge "numpy=1.26.4" "matplotlib=3.7.3" "scipy=1.14.1" "pandas=1.5.3" "seaborn=0.11.2" -y -q
#💬 Install PyQt6 required to use the GUI analyzer tool (gmx_MMPBSA_ana). Not needed for HPC
python -m pip install "pyqt6==6.7.1"
#如果没有gromacs可以用conda安装,如果用apt-get 之类的安装过就不需要了
conda install -c conda-forge "gromacs<=2023.4" pocl -y -q
# gmxMMPBSA安装
python -m pip install gmx_MMPBSA
2.使用
gmxMMPBSA官网其实写的很清楚,https://valdes-tresanco-ms.github.io/gmx_MMPBSA/dev/examples
这里简单介绍一下我经常用到的蛋白配体能量分解:
mpirun -np 6 gmx_MMPBSA -O -i mmpbsa.in -cs md_0_100.tpr -ct md_dt_100.xtc -ci index.ndx -cg 1 13 -cp topol.top -o FINAL_RESULTS_MMPBSA.dat -eo FINAL_RESULTS_MMPBSA.csv -do FINAL_DECOMP_MMPBSA.dat -deo FINAL_DECOMP_MMPBSA.csv
这里我采用的多线程版本
mmpbsa.in脚本如下:
Sample input file for decomposition analysis
This input file is meant to show only that gmx_MMPBSA works. Althought,
we tried to used the input files as recommended in the Amber manual,
some parameters have been changed to perform more expensive calculations
in a reasonable amount of time. Feel free to change the parameters
according to what is better for your system.
&general
sys_name="Decomposition",
startframe=1,
endframe=10,
forcefields="leaprc.protein.ff14SB"
/
&gb
igb=5, saltcon=0.150,
/
#make sure to include at least one residue from both the receptor
#and ligand in the print_res mask of the &decomp section.
#this requirement is automatically fulfilled when using the within keyword.
#http://archive.ambermd.org/201308/0075.html
&decomp
idecomp=2, dec_verbose=3,
print_res="within 4"
/
gromacs使用PCA绘制能量景观图
之前看到文献中有能量景观图,非常羡慕,而且感觉很高大上,等自己真正体会了主成分分析以后就比较释然了,但是若本身不是做模拟的,用来放一张图在支撑材料我感觉还是非常的好。
示例图如下:
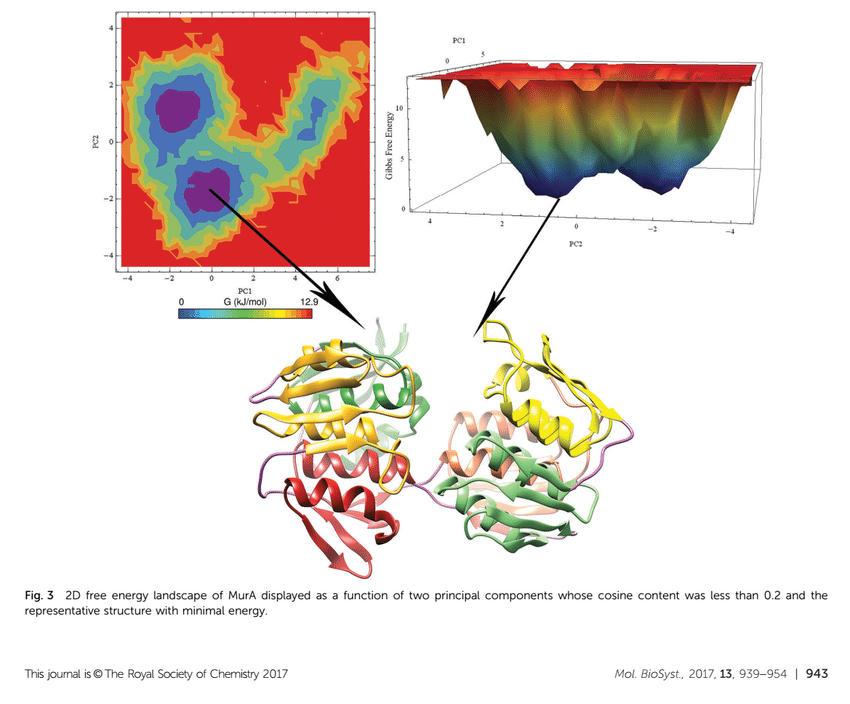
比对结构
Tip:
之前一直卡在后面计算时间很长,后来才发现是未进行蛋白比对的缘故,若没有进行蛋白比对(即蛋白不乱动),请先蛋白比对一下,命令如下:
gmx trjconv -f md.xtc -s md.tpr -fit rot+trans -o mdfit.xtc
生成协方差
首先我们需要使用gromacs covar来生成协方差矩阵并计算本征向量和本征值:
gmx covar -s md_0_1.gro -f md_0_100_fit.xtc -o eigenvalues.xvg -v eigenvectors.trr -xpma covapic.xpm
运行后会询问选择哪些原子范围做比对,选择C-alpha ,之后会询问选择哪些原子范围做PCA,选择C-alpha 或者其他均可。
最后会生成协方差的图形。但是这个最好在bio3d中,效果更佳,如下图所示:
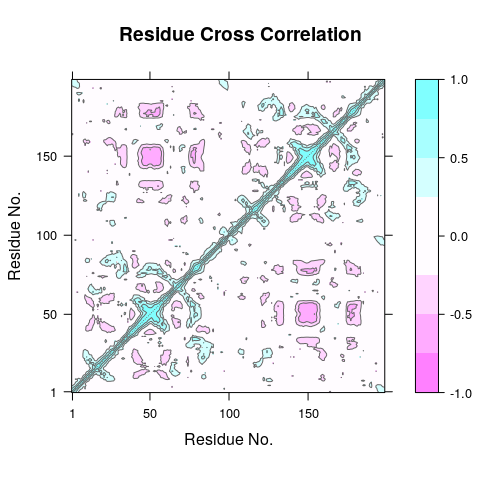
蓝色表示正相关,红色表示负相关,只需要看分界线的一面即可。
投影本征向量
若需要看PC1和PC2的投影,可以执行:
gmx anaeig -f md_0_100_fit.xtc -s md_0_1.gro -v eigenvectors.trr -first 1 -last 2 -2d 2d.xvg
同样bio3d上面可能好看那么一丢丢:
但是这样的缺点就是会丢失时间系数,所以我们只能一个PC一个PC的倒出:
gmx anaeig -f md_0_100_fit.xtc -s md_0_1.gro -v eigenvectors.trr -last 1 -proj pc1.xvg
gmx anaeig -f md_0_100_fit.xtc -s md_0_1.gro -v eigenvectors.trr -first 2 -last 2 -proj pc2.xvg
当然也可以写在一条里面。
再用sham.pl进行合并:
perl sham.pl -i1 pc1.xvg -i2 pc2.xvg -data 1 -data2 1 -o pc12_sham.xvg
此处也可以使用pc_combine.py脚本
python pc_combine.py pc1.xvg pc2.xvg pc12_sham.xvg
再使用sham工具计算Gibbs自由能:
gmx sham -f pc12_sham.xvg -ls FES.xpm
#或者可以设置温度等其他选项
gmx sham -tsham 310 -nlevels 100 -f pc12_sham.xvg -ls pc12_gibbs.xpm -g pc_12.log -lsh pc12_enthalpy.xpm -lss pc12_entropy.xpm
参数具体如下:
-tsham : 设定温度
-nlevels: 设定FEL的层次数量
-f : 读入组合文件
-ls : 输出自由能形貌图(Gibbs自由能)
-g :输出log文件
-lsh : 焓的形貌图
-lss : 熵的形貌图
生成的FES.xpm文件可以在linux系统下直接查看,但是不美观,我们可以使用xpm2png.py
转换为文本文件用其他软件做图:
xpm2png.py -ip yes -f gibbs.xpm
[注意该脚本为python2版本]
python2.7 xpm2txt.py -f FES.xpm -o free-energy-landscape.txt最后可以进行绘图,我是使用的matplotlib[注意我这里用的python3版本]:
# -*- coding:utf-8 -*-
#导包
import pandas as pd
import matplotlib.pyplot as plt
import numpy as np
# 读取文件
data=pd.read_csv('free-energy-landscape.txt',sep='\t',header=None)
fesvalue=np.array(data[2])
# 因为默认为32*32=1024,所以把其整为32*32格点,横纵坐标可抛弃,因为都是依次的,而且意义不大。
plotvalue=np.reshape(fesvalue,(32,-1))
# 绘图
plt.figure(figsize=(8,8))
plt.imshow(plotvalue,cmap='jet',interpolation='bilinear')
plt.xticks([],[])
plt.yticks([],[])
plt.xlabel('PC1',fontsize=16)
plt.ylabel('PC2',fontsize=16)
#plt.show()
plt.savefig('fes.png',dpi=600)
参考【不分先后】
参考1:https://www.mail-archive.com/gromacs.org_gmx-users@maillist.sys.kth.se/msg23365.html
参考2:http://sobereva.com/73
参考3:https://jerkwin.github.io/2017/10/20/GROMACS%E5%88%86%E5%AD%90%E5%8A%A8%E5%8A%9B%E5%AD%A6%E6%A8%A1%E6%8B%9F%E6%95%99%E7%A8%8B-%E5%A4%9A%E8%82%BD-%E8%9B%8B%E7%99%BD%E7%9B%B8%E4%BA%92%E4%BD%9C%E7%94%A8/
参考4:https://zhuanlan.zhihu.com/p/421494858